The medications we rely on to manage our health represent the culmination of a remarkable scientific odyssey. In this article series, we delve into the fascinating world of drug development, meticulously dissecting each stage of this intricate process. From the initial identification of promising molecules to the rigorous testing that paves the way for groundbreaking new treatments, we’ll explore the science that underpins the medications that improve countless lives. Prepare to embark on a journey of discovery and gain a deeper appreciation for the dedication and expertise that bring these life-changing advancements to fruition.
Stage 1: Discovery and Development – The Spark of Innovation
The first stage of drug development, often referred to as Discovery and Development, lays the groundwork for the entire process. It’s where the initial spark of a potential new drug is identified and nurtured. Here’s a breakdown of this crucial phase:
Target identification and validation:
The journey begins with a specific disease or condition. Researchers delve into the underlying biological processes to identify a target molecule – a protein, enzyme, or receptor – that plays a key role in the disease’s progression. This target becomes the focal point for drug development efforts.
There are several strategies for finding potential drug candidates:
- High-throughput screening (HTS): Testing libraries of existing compounds or natural products against the identified target molecule to see if they interact desirably.
- High-content screening (HCS): Involves the use of imaging and advanced automation to extract multi-parameter data describing phenotypic changes in biological systems.
- In Silico platforms: Computer-aided drug design (CADD) has become an integral part of drug discovery. It utilises computer modelling to design molecules with specific properties that could interact with the target molecule.
- Rational drug design: Leveraging existing knowledge about the target molecule and disease to design drugs that target specific mechanisms.
Lead Optimisation:
Once a promising candidate molecule is identified (a “lead”), researchers work to optimise its properties. This may involve:
- Improving potency: Increasing the drug’s ability to bind to the target molecule and produce a desired effect.
- Enhancing selectivity: Ensuring the drug interacts primarily with the target molecule and minimises unintended interactions with other molecules in the body.
- Optimising pharmacokinetics: Studying how the body absorbs, distributes, metabolises, and excretes (ADME) the drug to ensure it reaches its target site in sufficient concentrations.
Stage 2: Preclinical Research – Laying the Groundwork for Safety
The second stage of drug development, preclinical research, acts as a crucial bridge between the initial discovery phase and human testing. Here, potential drug candidates identified in the first stage undergo rigorous testing in a controlled laboratory environment to assess their safety and potential effectiveness before they are ever given to humans.
Key Aspects of Preclinical Research:
In Vitro Studies:
- Researchers use cell cultures to study how the drug interacts with specific cell types and evaluate its potential effects on cellular processes.
- These studies can provide insights into the drug’s mechanism of action (MOA), identify potential off-target effects, and assess its potential cytotoxicity (toxicity to cells).
**Checkout these solutions from Atlantis Bioscience for your in vitro studies: 3D solutions, microfluidics, primary hepatocyes, biomimetic membranes
In Vivo Studies:
- Animal models, such as mice, rats, or rabbits, are used to evaluate the drug’s overall effects on a living organism. This allows researchers to assess the drug’s pharmacokinetics (ADME) and its potential impact on various organ systems.
- Animal models provide valuable information about the drug’s potential toxicity, efficacy in a whole organism, and optimal dosing strategies.
**Checkout these solutions from Atlantis Bioscience for your in vitro studies : Mouse models, preclinical testing services
Importance of Preclinical Research:
- Safety First: Preclinical research serves as a vital safety screen. By identifying potential toxic effects or unintended interactions in cells and animals, researchers can eliminate unsafe drug candidates before they reach human trials.
- Optimising Development: Preclinical studies provide valuable data to guide further drug development. This information helps researchers refine the drug’s formulation, dosage, and administration schedule for future clinical trials.
- Understanding Mechanism of Action: By observing the drug’s effects on cells and animals, researchers gain a deeper understanding of how it works at the molecular and physiological level.
Ethical Considerations:
Researchers have an ethical obligation to minimise animal suffering during preclinical research. Strict guidelines are in place to ensure humane treatment of animals used in drug development. The use of animal models has limitations, and their responses to drugs may not always translate perfectly to humans. However, they remain an essential tool in the early stages of drug development.
Stage 3: Clinical Research – Testing in Humans: The Defining Phase
Clinical research, the third stage of drug development, marks the first time a potential medication is tested in humans. This phase is crucial for determining the drug’s safety, efficacy, and optimal dosage for treating a specific disease. Here’s a breakdown of what clinical research entails:
Rigorous and Ethical:
Clinical research is a highly regulated and ethically conducted process. Strict guidelines and oversight by regulatory bodies like the FDA (Food and Drug Administration) ensure the safety and well-being of participants.
The Investigational New Drug Process:
Drug developers, or sponsors, must submit an Investigational New Drug (IND) application to FDA to obtain authorisation for administering an investigational drug to humans before beginning clinical research. An IND application typically includes information on the animal pharmacology and toxicology studies, manufacturing information and clinical protocols. By submitting a well-prepared IND application, sponsors can gain FDA approval to proceed with clinical trials, a critical step in developing safe and effective new drugs.
Phased Approach:
Clinical research typically follows a phased approach, with each phase building upon the findings of the previous one:
- Phase I: A small group of healthy volunteers or people (typically 20-100) with the disease/condition are administered the drug to assess its safety, tolerability, and pharmacokinetics (how the body absorbs, distributes, metabolises, and excretes the drug).
- Phase II: The drug is tested in a larger group of people (up to several hundred) with the disease/condition. This phase focuses on evaluating the drug’s efficacy (effectiveness) for treating the disease and identifying common side effects.
- Phase III: The most extensive phase, involving hundreds or even thousands of patients across multiple clinical sites. This phase provides most of the safety data as these studies are larger and longer in duration, and therefore the results are more likely to show long-term or rare side effects
- Phase IV: Also known as post-marketing surveillance, this phase occurs after the drug is approved and commercially available. It monitors the drug’s long-term safety and effectiveness in a broader population under real-world conditions.
Informed Consent:
Participation in clinical research is voluntary. Patients must undergo a rigorous informed consent process, where they receive a detailed explanation of the study’s objectives, potential risks and benefits, and their rights as participants.
Importance of Clinical Research:
- Evidence-Based Medicine: Clinical research provides the strongest evidence for the safety and efficacy of new drugs. It allows researchers to compare different treatments and identify the most effective options for patients.
- Individualised Medicine: Clinical trials can help identify subgroups of patients who may respond best to a particular drug, paving the way for personalised medicine approaches.
- Advancing Medical Knowledge: Clinical research contributes significantly to our understanding of diseases and treatment mechanisms, leading to continuous advancements in healthcare.
Challenges of Clinical Research:
- Cost and Time: Clinical research is a lengthy and expensive undertaking.
- Recruitment: Finding and enrolling eligible participants can be challenging, especially for rare diseases.
- Placebo Effect: In some cases, patients receiving a placebo (a look-alike inactive substance) may experience improvement due to the power of suggestion, making it difficult to assess the true effectiveness of the drug.
Stage 4: FDA Review – Scrutinising for Safety and Efficacy
The New Drug Application (NDA) is the final hurdle a new drug needs to clear before it can be legally marketed and prescribed to patients. Submitted to the FDA, an NDA acts as a formal proposal from a drug sponsor (pharmaceutical company, researcher) requesting approval to sell their drug for specific medical conditions. The FDA rigorously reviews NDAs, a lengthy process taking months to even years, as the FDA meticulously ensures the drug meets their stringent safety and efficacy standards.
The Role of the FDA:
The FDA serves as the gatekeeper, ensuring the safety and efficacy of new drugs before they reach the public. Their review process involves a thorough examination of all the data collected throughout drug development:
- Preclinical research data: Information on the drug’s effects in cell cultures and animal models.
- Clinical trial data: Detailed reports on the design, conduct, and results of all clinical trials, including data on safety, efficacy, and side effects.
- Manufacturing information: Data on the drug’s manufacturing process, quality control measures, and potential risks associated with manufacturing.
- Labelling: Proposed labelling for the drug, which includes information about its uses, dosage, potential side effects, and warnings.
A Team Effort:
The FDA review process involves a team of experts, including physicians, pharmacologists, chemists, statisticians, and other specialists. Each team member meticulously evaluates the submitted data to ensure the drug meets the following criteria:
- Safety: The drug’s benefits outweigh the potential risks for the intended patient population.
- Efficacy: The drug is demonstrably effective in treating the target disease.
- Quality: The drug is manufactured consistently according to established quality standards.
- Labelling: The drug’s labelling accurately reflects its uses, risks, and benefits.
Possible Outcomes:
The FDA review process can lead to several outcomes:
- Approval: If the drug meets all the requirements, the FDA grants marketing approval, allowing the manufacturer to sell the drug commercially.
- Request for Additional Information (RAI): The FDA may request additional data or clarification on specific aspects of the drug’s development before making a final decision.
- Not Approved: If the FDA determines that the drug does not meet its safety or efficacy standards, it will not be approved for marketing.
Stage 5: FDA Post-Market Safety Monitoring – Vigilance After Approval
The journey of a drug doesn’t end with FDA approval. The fifth and final stage of drug development, FDA post-market safety monitoring, ensures continued vigilance even after a medication becomes commercially available. Here’s why this ongoing process is crucial:
Why Monitor After Approval?
Clinical trials, while thorough, can involve a limited number of participants and may not capture all potential side effects, especially rare ones. Additionally, long-term effects and interactions with other medications might not be fully evident during clinical trials.
FDA’s Active Role:
The FDA maintains a robust post-market safety monitoring program to identify any unforeseen safety issues with approved drugs. This program relies on several key elements:
- Adverse Event Reporting System (FAERS): This database allows healthcare professionals, patients, and consumers to report any adverse events (negative side effects) experienced while using the drug.
- Risk Evaluation and Mitigation Strategies (REMS): For high-risk drugs, the FDA may require REMS programs to ensure safe use. These programs might involve mandatory education for prescribers, patient registries, or special dispensing limitations.
Benefits of Post-Market Monitoring:
- Early Detection of Safety Issues: By actively collecting data on adverse events, the FDA can identify any unexpected safety concerns early on, potentially preventing serious harm to patients.
- Refining Drug Labels: Based on data from post-market monitoring, the FDA can update drug labels to include information about newly discovered side effects or potential interactions with other medications.
- Informing Future Drug Development: Data from post-market surveillance can inform the development of safer and more effective drugs in the future.
Shared Responsibility:
While the FDA plays a central role in post-market surveillance, healthcare professionals and patients also have a responsibility to report any adverse events experienced with approved medications. This reporting helps create a more comprehensive picture of the drug’s safety profile.
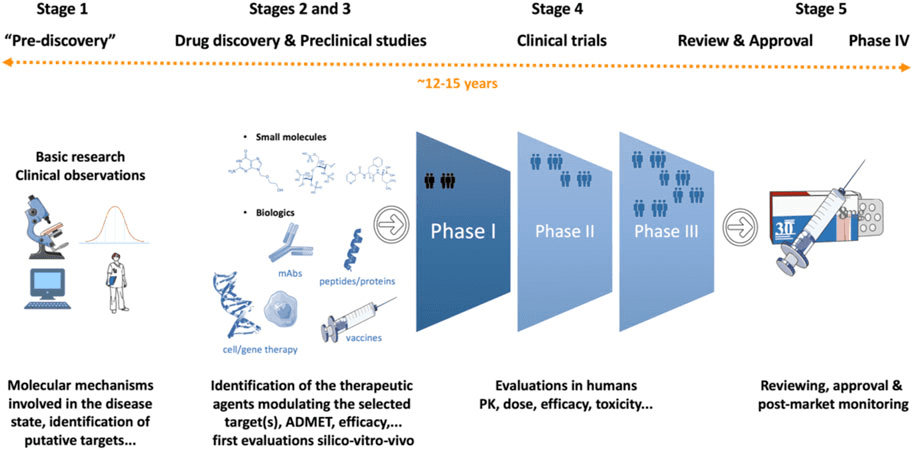
Figure 1: Drug discovery and development process.
Credit: Singh N., Vayer P., et.al., doi: 10.3389/fddsv.2023.1201419.
Reproduced under the Creative Commons license
Concluding Remarks
The journey from a scientific hunch to a life-saving medication is a long and intricate one. Discovering and bringing a new drug to market typically takes 12-15 years and costs approximately $2.8 billion USD. These staggering numbers highlight the immense dedication, expertise, and resources required to translate scientific innovation into tangible benefits for patients.
Today’s competitive landscape throughout the drug development process demands a delicate balance. Research scientists strive to apply rigorous qualitative and quantitative analyses to bring lead drug candidates to market in the shortest timeframe possible. However, this pursuit of efficiency must never compromise the meticulous testing and safety evaluations that are fundamental to ensuring the well-being of patients.
As we move forward, exciting advancements in technology and research methodologies hold the potential to streamline the drug development process while maintaining the highest safety standards. By fostering collaboration and innovation, we can accelerate the discovery and delivery of life-changing medications to patients in need.
References:
Chang Y, Hawkins BA, Du JJ, Groundwater PW, Hibbs DE, Lai F. A Guide to In Silico Drug Design. Pharmaceutics. 2022 Dec 23;15(1):49. doi: 10.3390/pharmaceutics15010049.
Savoji H, Mohammadi MH, Rafatian N, et al. Cardiovascular disease models: A game changing paradigm in drug discovery and screening. Biomaterials. 2019;198:3-26. doi:10.1016/j.biomaterials.2018.09.036
Singh N, Vayer P, Tanwar S, Poyet J, Tsaioun K, Villoutreix BO. Drug discovery and development: introduction to the general public and patient groups. Frontiers in Drug Discovery. 2023; (3). doi:10.3389/fddsv.2023.1201419